
{kind=link}
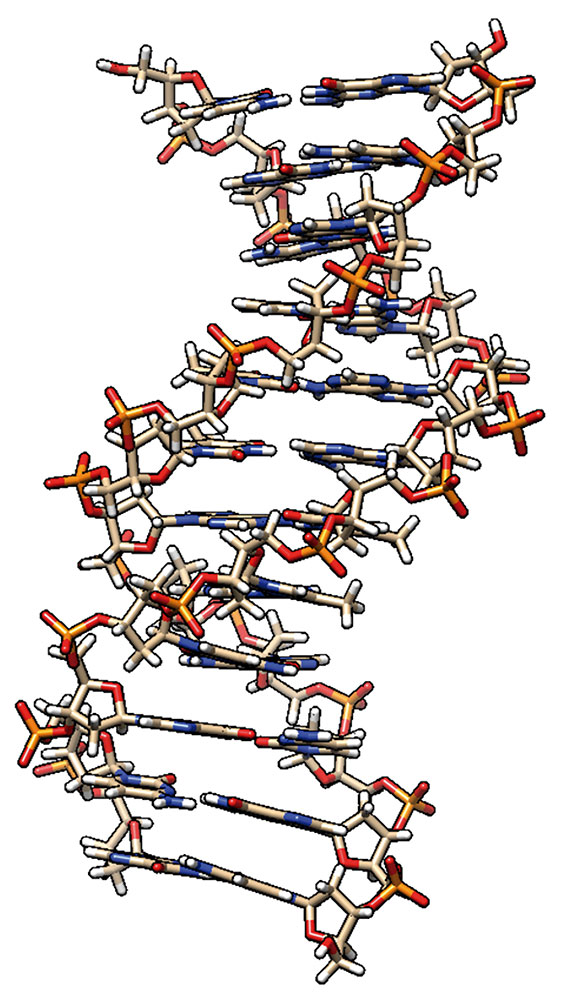
La estructura espacial del ácido desoxirribonucleico (ADN) codifica toda la información necesaria para el desarrollo individual y la reproducción de los organismos vivos. El descubrimiento de esta estructura en 1953 por Watson y Crick proporcionó, hasta ese momento y en principio, una respuesta a la pregunta de cómo, desde el punto de vista de la física, pueden existir, reproducirse y desarrollarse los sistemas vivos. A partir del modelo que construyeron Watson y Crick: el ADN contiene dos cadenas polinucleotídicas complementarias antiparalelas, con cuatro tipos de bases nitrogenadas, dos purinas (adenina y guanina) y dos pirimidinas (timina y citosina), y fragmentos repetidos de un esqueleto de azúcar-fosfato químicamente homogéneo, se dedujeron, en términos generales, los mecanismos del almacenamiento, reproducción, uso y cambios evolutivos del material genético (Figura 1). Desde entonces, se han publicado miles de trabajos para investigar este biopolímero, utilizando una amplia gama de métodos experimentales y teóricos. Las capacidades modernas de los métodos físicos y las tecnologías de la información han hecho posible crear un banco de datos de las coordenadas de más de 10 mil estructuras de fragmentos de ADN y sus complejos con otras moléculas biológicamente importantes. Este banco está en constante crecimiento, siendo útil para el análisis comparativo de las características estructurales de los fragmentos y para la investigación de los mecanismos del funcionamiento del ADN, lo que permite realizar nuevas generalizaciones cada vez más interesantes sobre la estructura y el funcionamiento de la Molécula Principal de la Vida (Figura 1).
Para esta investigación se utilizan todos los métodos disponibles para modelar la estructura molecular, desde la construcción manual de fragmentos moleculares a partir de modelos plásticos de átomos y enlaces, (cómo se descubrió realmente la estructura de la doble hélice y de estructuras macromoleculares proteicas) hasta los métodos más modernos de la mecánica cuántica, cuyo uso actualmente sólo es posible en fragmentos relativamente pequeños (cientos de átomos) en principio, independientemente del software informático.
La elección del método depende de la tarea en cuestión, por ejemplo, si queremos confirmar, o describir con mayor detalle nuevas estructuras conocidas, o hacer una nueva predicción, cuyos resultados se utilizarán en futuros trabajos. En este último caso, no se puede prescindir de los cálculos de la mecánica cuántica, disponibles con diversos grados de complejidad y utilizando diferentes paquetes de software. Todo esto sin perder de vista la limitación fundamental de estos métodos teóricos, que todos ellos requieren del uso de supercomputadoras y muchas horas de cálculo. La comparación de los valores obtenidos durante el modelado molecular con los datos experimentales es el principal criterio para valorar la exactitud de las conclusiones obtenidas.
El método utilizado en muchos de los estudios de ADN es el método de la Mecánica Molecular. Este método simple y “mecanicista” puede considerarse como el desarrollo de una descripción cuantitativa para representar la estructura espacial de una molécula en química orgánica en forma de átomos y enlaces químicos. En este caso, la molécula es representada por un conjunto de átomos que interactúan según las leyes simples de la física clásica (o semiclásica), como la ley de Coulomb para la interacción de cargas en los átomos o la ley de Hooke para el cambio de la energía en el enlace químico. La principal aproximación de este método es la aditividad de la energía de interacción entre pares de átomos y la diferente naturaleza física las interacciones, cuya suma es la energía total de una molécula o de un complejo de moléculas con una determinada disposición espacial de sus átomos. El uso de los métodos para buscar mínimos de energía nos permite encontrar las posiciones mutuas energéticamente más favorables de los átomos, es decir, las conformaciones más probables de una molécula o complejo.
La justificación para considerar a los sistemas biomoleculares de esta forma nos la da la mecánica cuántica, a través de la aproximación de Born-Oppenheimer, que permite la separación de los grados de libertad de los electrones y núcleos del sistema. Ningún método mecánico-cuántico para estudiar moléculas puede prescindir de esta aproximación. A pesar de una descripción clásica y tan poco rigurosa y en muchos casos formalmente incorrecta de un sistema cuántico, cuando se aplica correctamente a ciertos sistemas moleculares, no solo da resultados consistentes con el experimento, sino que también permite hacer predicciones antes de realizar el experimento (y a menudo en lugar de éste). La clave para la obtención de resultados correctos es la elección de los parámetros en las expresiones matemáticas de la energía de una molécula o complejo de moléculas, basados en la comparación con datos experimentales. Estas expresiones, junto con los parámetros seleccionados, se denominan “campo de fuerzas”. Actualmente existe una amplia variedad de campos de fuerzas que se han aplicado con éxito en el estudio de los ácidos nucleicos y sus complejos.
Mucho antes del desarrollo de los programas informáticos y de los campos de fuerza universales de la mecánica molecular, con el uso de los métodos mecanicistas simplificados, se pudo hacer predicciones interesantes sobre las propiedades de la molécula de ADN y los mecanismos para realizar sus funciones biológicas. Entre ellas, sobre la labilidad de la doble hélice, es decir la posibilidad de cambiar sus parámetros geométricos y su dependencia con la secuencia de nucleótidos, la cual se predijo varios años antes de que aparecieran los primeros trabajos experimentales (análisis de difracción de rayos X) que lo demostraron. La posibilidad de adaptar la geometría del dúplex de ADN a diferentes pares de bases erróneos (que no son de Watson y Crick), dando lugar a mutaciones como base de la evolución biológica, esto se predijo varios meses antes de que se descifrara experimentalmente la primera estructura de este tipo.
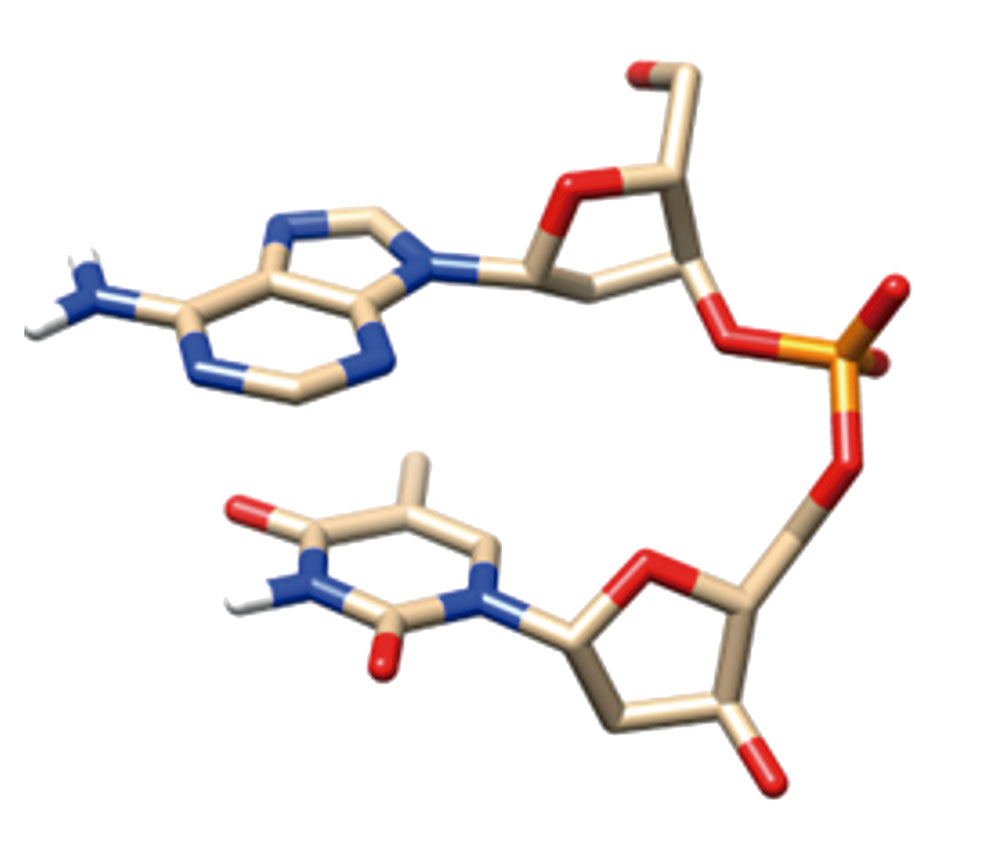
Los datos experimentales acumulados de una gran cantidad de estructuras de fragmentos de ADN conteniendo varias decenas de pares de nucleótidos han planteado una serie de interrogantes, cuya solución es imposible sin los modelos computacionales. Entre estas tareas se encuentra la de evaluar la contribución en la formación y funcionamiento de ADN respecto de sus subunidades individuales (bases, residuos de azúcar, grupo fosfato) y sus interacciones en los fragmentos mínimos (par de bases, nucleósidos, nucleótidos, fragmento de cadena azúcar-fosfato, fragmentos mínimos de cadena simple o espiral doble). Uno de los resultados más importantes de nuestra investigación de los últimos veinte años es la suposición y luego la fundamentación de la conclusión obtenida después de investigar muchos patrones generales de formación de la estructura espacial de un dúplex de ADN, incluyendo la dependencia de la estructura espacial de la secuencia de nucleótidos, están determinadas por las interacciones de las subunidades en un fragmento mínimo de cadena única (dos nucleósidos, conectados por un grupo fosfato (Figura 2)). Muchas conformaciones de dicho fragmento, incluidas las formas A y B del ADN, conocidas desde los primeros años después del descubrimiento de la doble hélice, corresponden a los mínimos de energía de este fragmento y del fragmento de cadena azúcar-fosfato. Esta conclusión, que inicialmente hicimos con recursos informáticos limitados para las conformaciones B con métodos DFT de la mecánica cuántica, se confirmó luego en mayor cantidad de estructuras y con cálculos más precisos de la mecánica cuántica en colaboración con investigadores de otros países. Muchos de los resultados (pero no en todos los casos) pueden reproducirse con los métodos modernos de la mecánica molecular.
Otro resultado importante de estos estudios es el reconocimiento del papel esencial de la estructura de la cadena azúcar-fosfato en la formación del dúplex de Watson-Crick, la cual depende de la secuencia de bases. Un patrón que se observó es el comportamiento de las bases nucleicas cuando se apilan en el dúplex de ADN, se producen ciertos patrones característicos sobre el apantallamiento de las bases apiladas. A pesar de los avances que se han logrado aún hay todavía varias preguntas sin contestar sobre el papel biológico de las diferentes clases conformacionales y al papel de los patrones de formación de estructuras espaciales descubiertos y, quizás, aún no descubiertos, en ciertas regiones de la macromolécula de ADN. La investigación continua.
* [email protected], [email protected], [email protected]